


《自然》子刊:蛋白質結構預測新算法可大幅提高預測效率
10月9日,國際頂級學術期刊《自然》旗下子刊《機器智能》發表了百度飛槳螺旋槳聯合百圖生科研發的文心生物計算大模型的一項成果,在這一模型中,由于創新了人工智能的訓練方案,蛋白質結構預測時間被大幅縮短。
該論文顯示,當前蛋白質結構預測的人工智能大模型,如AlphaFold2(阿爾法折疊2),在預測蛋白質結構之前需要做一個前置工作,就是搜索蛋白的同源進化信息,這一工作比較耗時,需要幾十分鐘甚至更久。
為了在準確的前提下提高預測效率,文心生物計算大模型研發團隊提出了全新的算法訓練方法,他們首次利用自監督學習范式,通過3億數據預訓練了一個具有數千萬個初級結構的大規模蛋白質語言模型。
“自監督學習獲得的模型與AlphaFold2的基本組件相結合,可以將此前的耗時環節直接省略掉。”研發人員介紹,由于預先訓練了蛋白質語言模型,人工智能在預測前已經掌握了蛋白質的構象規則,因此無需再學習同源蛋白的進化信息,就可以直接從一級序列預測三維結構。
論文還對文心生物計算大模型的這一算法新策略進行了驗證。以門蛋白7et2_H(蛋白長度697)的結構預測任務為例,用AlphaFold2預測其結構需要1280秒(超過21分鐘),而文心的新算法策略只需要11秒就完成了任務,速度提高115倍。
全新的算法策略不僅能更好適配到蛋白設計、大規模虛擬篩選等需要頻繁預測蛋白結構的任務中,且在多肽、抗體、納米抗體等與大分子藥物設計更相關的高可變蛋白場景上,效果也較優。
這一算法還被應用于業界公認的抗原抗體匹配的任務中,為新冠病毒的刺突蛋白準確預測了抗原抗體對接面,預測的復合體構象與真實實驗值的重合度高于主流蛋白結構預測模型(見下圖)。
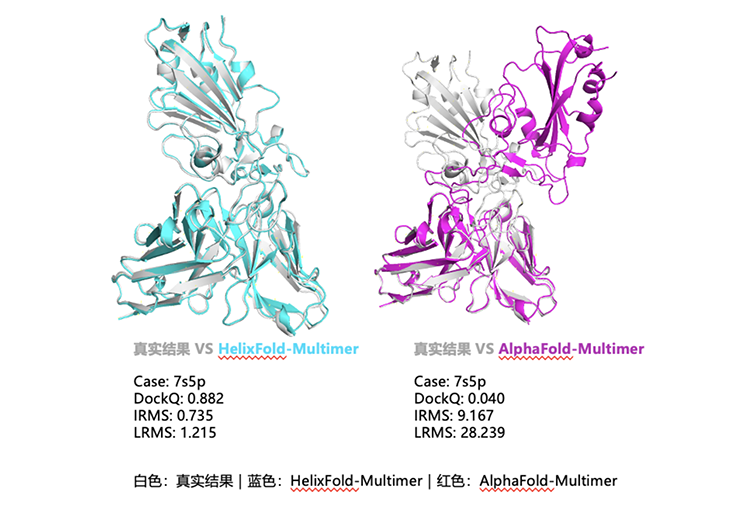
據介紹,該模型已經落地國家超算成都中心,通過超算平臺賦能川渝地區蛋白領域的科學研究項目。此外,目前已經應用于多肽藥物設計,驗證多肽藥物設計有效性等方面,助力更高效的蛋白分析,以提高大分子創新藥的探索效率。
(研發團隊供圖)


